
Compte-rendu du 9° Symposium international du syndrome de Marfan qui s’est déroulé à Paris du 25 au 27 septembre 2014
Avec l’aimable autorisation de l’AFSMa
Back to top
Présentation par le Dr Jean Bachet
Décrit en 1896 par Antonin Marfan, pédiatre à l’Hôpital des Enfants Malades et Professeur à la Faculté de Médecine de Paris, le syndrome qui porte son nom est une maladie du tissu conjonctif (tissu de soutien de tous les tissus de l’organisme) d’origine génétique.
Il est généralement dû à des mutations du gène FBN1 placé sur le chromosome 21 (15q21) qui code pour la fibrilline-1, une protéine essentielle du tissu conjonctif.
La survenue du syndrome est estimée à environ une naissance sur 5000.
La maladie est dite « autosomique » et peut donc toucher indifféremment les deux sexes.
Le syndrome associe des combinaisons variables d’atteintes squelettiques et musculaires, cardio-vasculaires, ophtalmologiques et pulmonaires.
Les signes de la maladie peuvent apparaître à des âges très variables mais sont généralement découverts à l’adolescence. Ils sont eux-mêmes très variables d’un sujet à l’autre y compris à l’intérieur d’une même famille.
L’attention est généralement attirée, soit du fait de l’existence d’autres cas familiaux, soit par les anomalies squelettiques (taille anormalement grande, grande longueur des bras, doigts anormalement longs –arachnodactylie- très grande laxité ligamentaire, etc.).
L’atteinte ophtalmique entraine généralement un myopie sévère par luxation du cristallin.
Mais ce sont surtout les risques cardiovasculaires qui dominent la maladie et conditionnent son pronostic. En effet, du fait de la fragilité du tissu conjonctif, l’aorte a tendance à se dilater progressivement, en particulier dans sa partie toute initiale à la sortie du cœur et peut, soit se rompre (ce qui est rare) soit, plus fréquemment, se « disséquer », c’est à dire se déchirer partiellement longitudinalement ce qui peut rapidement entrainer le décès par saignement dans le péricarde et compression majeure du cœur (tamponnade), et/ou des « mal perfusions » coronariennes, cérébrales, viscérales ou des membres. La mortalité spontanée de cet accident est estimée à environ 50% dans les 48 premières heures. Il s’agit donc d’une urgence chirurgicale majeure.
Actuellement, du fait de la prise en charge précoce, de la surveillance régulière et de la chirurgie préventive des anomalies cardio-vasculaires, l’espérance de vie des sujets atteints est proche de celle de la population générale.
Dans de nombreux pays occidentaux, les malades atteints du syndrome de Marfan se sont regroupés en associations actives qui ont grandement participé à la connaissance et à la prise en charge médicale et sociale des patients.
En France, l’Association Française des Syndromes de Marfan et Associés a été créée en 1990. Elle fait partie de l’Association des Maladies Rares dans laquelle elle a une place tout à fait essentielle.
C’est à travers l’expérience chirurgicale du Service de Chirurgie Cardio-vasculaire de l’hôpital Foch que l’ADETEC a été mise en contact avec l’AFSM et a décidé de lui apporter un soutien, certes modeste, mais constant et fraternel.
Nous tenons à remercier les Présidents de l’ASFMa et de la Fondation Marfan de l’autorisation qu’ils nous ont donnée de reproduire ici l’essentiel du compte-rendu du 9° Symposium de ces organismes .
Back to top
Compte rendu du 9eme Symposium international du syndrome de Marfan
Le symposium international du syndrome de Marfan et apparentés s'est déroulé du 25 au 27 septembre 2014 à Paris. Cette 9ème édition a remporté un énorme succès avec un record de participation des chercheurs qui sont venus du monde entier. L'objectif de ce congrès a été de présenter les nouvelles avancées scientifiques en matière de compréhension, de gestion et de traitement de la maladie. Un effort particulier a été réalisé pour intégrer à la fois des conférences portant sur des recherches cliniques et sur des recherches fondamentales.
Session 1 : Etude sur le Losartan
Historique :
En 2006, Habashí et ses collaborateurs ont démontré que le Losartan à forte dose permettait de diminuer la vitesse de dilatation de l'aorte ascendante chez un modèle de souris atteintes du syndrome de Marfan.
Depuis, plusieurs études cliniques chez l’homme ont été développées afin d'étudier l'efficacité du Losartan chez les patients atteints du syndrome de Marfan.
Etude Losartan sans groupe contrôle
En 2008, Brooke et ses collaborateurs ont démontré dans une cohorte pédiatrique Marfan que le Losartan permettrait de diminuer progressivement la dilation de l’aorte ascendante. En 2013. Pees et ses collaborateurs ont également publié une étude, réalisée sur une cohorte de jeunes Marfan, où le Losartan conduisait à une amélioration au niveau de tous les segments proximaux de l'aorte.
Etude de la combinaison Losartan + ß bloquants vs ß bloquants
En 2013, l'étude pédiatrique taïwanaise a révélé que la combinaison du Losartan + ß bloquants serait plus efficace que les ß bloquants seuls dans la progression de la dilatation de l'aorte ascendante.
Etude du Losartan vs ß bloquants
En 2014, Mueller et ses collaborateurs ont observé, dans une plus grande cohorte de jeunes Marfan, un effet similaire entre le Losartan et les ß bloquants.
Etude du Losartan vs placebo
L'étude clinique COMPARE (Pays-Bas), dont les résultats ont été publiés en 2013, démontre dans une cohorte d'adultes atteints du syndrome de Marfan, que le Losartan permettrait de diminuer la dilatation de l'aorte ascendante.
L’étude SARTAN réalisée en France est terminée et les résultats finaux seront prochainement publiés. Cette étude est :
- multicentrique (étude menée dans différents centres en France
- randomisée (les patients ont reçu au hasard le Losartan ou un placebo)
- double aveugle (ni le médecin, ni le patient, ne connaissait le traitement reçu)
- versus placebo (le placebo est un médicament qui ne contient aucun principe actif)
Cette étude a nécessité l'inclusion de 300 patients atteints du syndrome de Marfan selon les critères de Ghent. Ce type d'essai clinique a pour objectif de réellement déterminer l'efficacité du traitement. Les critères qui ont permis aux scientifiques de juger d'un bénéfice potentiel étaient l'évolution du diamètre aortique et la survenue d'événements tels que la dissection aortique. Cette étude n'a pas permis de démontrer une efficacité du Losartan sur l'évolution du diamètre de l'aorte, ainsi que sur la survenue d'évènements.
Une méta-analyse, c'est-à-dire une étude globale incluant les différentes études de plusieurs pays, est prévue afin de mieux éclaircir le bénéfice potentiel des sartans.
Session 2 : Avancée scientifique dans le domaine de la chirurgie aortique

Comparaison de deux techniques pour le remplacement de la racine aortique V-SARR versus CVG
L'objectif de cette étude a été de comparer les résultats obtenus après remplacement de la racine aortique selon deux méthodes : V-SARR (conservation de la valve aortique) et CVG (valve aortique artificielle).
316 patients atteints du syndrome de Marfan ont été inclus dans cette étude entre 2005 et 2010 dans différents centres d'étude. 239 patients (76%) ont été opérés selon la méthode V-SARR, et 77 (24%) selon la méthode CVG.
Lorsque l'on compare ces deux techniques 1 an après l'opération, aucune différence n'est observée pour la mortalité et les effets secondaires majeurs. En revanche, 7% des patients opérés avec la méthode V-SARR ont eu une insuffisance aortique, contre 0% avec la méthode CVG. Un suivi des patients à plus long terme sera nécessaire afin de réellement pouvoir comparer ces deux méthodes.
Indication pour la réparation aortique endovasculaire chez les patients atteints d'anévrisme de l'aorte thoracique .
Ces dernières années, le remplacement prophylactique de la racine aortique a permis d'augmenter l’espérance de vie des patients atteints du syndrome de Marfan.
Conséquence: cela a conduit à une forte augmentation des interventions au niveau de l'aorte ascendante et de l'aorte thoracique. Les données actuelles suggèrent que, dans ces circonstances, l'utilisation de la réparation aortique et vasculaire est une méthode faisable et sans danger. Bien qu'elle ne soit pas la stratégie de référence, elle permet d'obtenir des résultats acceptables chez certains patients à moyen terme.
Médecine personnalisée pour le remplacement de la racine aortique : la méthode PEARS
Les travaux du Dr Treasure récemment publiés dans le journal scientifique HEART en 2014, ont permis une grande avancée scientifique pour le traitement du syndrome de Marfan.
Le principe de cette technique innovante est de pouvoir reproduire la forme exacte de la racine aortique pour chaque patient et de créer un support physique qui va venir parfaitement épouser sa forme. L'avantage de cette méthode est la conservation de la valve aortique et de son architecture naturelle. Dans cette étude, 30 patients inclus entre 2004 et 2011 ont reçu cette intervention chirurgicale. Aucun des événements suivants n'ont été retrouvés chez ces patients : mortalité post-opératoire, ré-intervention sur la racine aortique, endocardite et maladie thromboembolique. Cette médecine personnalisée semble très prometteuse et a déjà reçu l'approbation du British National Institute for Health and Care Excellence.
Session 3 : Avancée scientifique dans le domaine de l'ophtalmologie
Ectopie du cristallin et syndrome de Marfan
Cette étude belge avait pour objectif de caractériser les atteintes oculaires de type ectopie du cristallin dans le syndrome de Marfan. Elle a permis de démontrer que 80% des patients atteints du syndrome de Marfan ont un certain degré d'ectopie du cristallin et ont également, pour la majorité, de la myopie. Une augmentation de la longueur axiale et une cornée plate sont retrouvées chez un peu moins de 30% des patients.
Les mutations présentes dans la fibrilline 1 sont responsables de 75% des ectopies du cristallin aussi bien pour les patients atteints du syndrome de Marfan que pour ceux atteints d'ectopie du cristallin isolée. Cela démontre que les analyses moléculaires sont indispensables pour un bon diagnostic.
Etude de corrélation génotype/phénotype dans une population de taïwanais atteints du syndrome de Marfan
Cette étude avait pour objectif d`établir une corrélation entre le génotype (mutation de la fibrilline 1) et le phénotype (manifestations cliniques) dans une population de Taïwanais atteints du syndrome de Marfan. Entre 2007 et 2013, 125 patients atteints du syndrome de Marfan avec une mutation de la fibrilline 1 ont été inclus dans cette étude. 85 mutations différentes ont été retrouvées dont la plupart localisés entre les exons 11 et 24 (28%), les exons 25-40 (22%), dans le «domaine cbEGF›› (63%) et dans le domaine TGFBP (19%).
Cette étude a permis de démontrer que les mutations présentes entre les exons 11 et 24 sont associées à des manifestations oculaires majeures. Celles retrouvées entre les exons 41-57 conduisent principalement à des
manifestations cardiovasculaires majeures et à des atteintes du squelette.
Ces travaux ont permis de montrer l'importance d'établir des liens entre le diagnostic génétique et les manifestations cliniques, bien que cela soit extrêmement difficile en raison de la variabilité des signes cliniques.
Session 4 : Atteintes osseuses et syndrome de Marfan
Croissance du squelette chez les enfants atteints du syndrome de Marfan
Cette étude a été menée en France afin de mieux comprendre l'évolution de la croissance du squelette chez les enfants atteints du syndrome de Marfan. Elle a nécessité l'inclusion de 259 enfants atteints du syndrome de Marfan avec une mutation dans le gène codant pour la fibrilline 1. Les enfants Marfan, filles ou garçons, ont tous une taille supérieure à la normale indépendamment de l'âge. La moyenne d'âge pour le début de puberté est significativement plus faible pour les enfants Marfan comparée à la normale (10 ans pour les filles, et 12 ans pour les garçons). La prévalence des manifestations cliniques au niveau du squelette varie avec l'âge. Par exemple, les déformations du pectus augmentent de 43% à 62% de l'âge de 0-6 ans à 15-17 ans. ll existe une grande variabilité des manifestions cliniques au niveau du squelette, mais la taille semble être un premier critère de diagnostic pour le syndrome de Marfan dans la population générale. Il serait intéressant de pouvoir établir une courbe standard de croissance pour les enfants Marfan afin d'essayer de prédire la taille définitive d'un enfant atteint.
La dysplasie acromélique est causée par des mutations dans FBN1, ADAMTS10, et ADAMTSL2
La dysplasie acromélique désigne un groupe de maladies qui est composé du syndrome de Weill-Marchesani, de la dysplasie geleophysique, de l'acromicrie et du syndrome de Myhre. Elle est caractérisée par une petite taille, des petits membres et une mobilité des articulations diminuée.
Des mutations dans le gène codant, la fibrilline 1, ont été retrouvées pour la forme dominante du syndrome de Weill-Marchesani, et des mutations dans le gène codant pour ADAMTS10 pour la forme récessive.
Des mutations dans le gène codant pour ADAMTSL2 ont été identifiées pour la forme récessive de la dysplasie geleophysique. Pour la forme dominante de la dysplasie geleophysique et pour l'acromicrie, des mutations dans les exons 41-42 de la fibrilline 1 ont été retrouvées.
Ces travaux suggèrent que ces mutations peuvent conduire à une dérégulation dans la voie de signalisation du TGF-B qui est essentielle pour le développement osseux.
Phénotype osseux chez les enfants et les jeunes adultes atteints du syndrome de Marfan
L'objectif de cette étude a été d'évaluer la densité minérale osseuse et les marqueurs du remodelage osseux chez les jeunes Marfan. Elle a été réalisée à partir de 44 enfants et de 18 jeunes adultes répondant aux critères de Ghent. Les marqueurs du remodelage osseux n'étaient pas modifiés comparés à la population normale. En revanche, les patients atteints du syndrome de Marfan avait une densité minérale osseuse au niveau du rachis lombaire significativement diminuée. L'évaluation de la masse osseuse semble donc être importante pour le suivi des enfants atteints du syndrome de Marfan.
Session 5 : Avancées scientifiques dans le domaine de la recherche cardiovasculaire dans les syndromes de Marfan et apparentés
Une nouvelle méthode pour détecter la dilatation aortique :
Pour évaluer la sévérité de dilatation de l'aorte thoracique, il est nécessaire de connaître quelle est la définition d'un diamètre de l`aorte normal. Grâce à un modèle mathématique reposant sur l'âge, le sexe et la taille, il est maintenant possible de déterminer une valeur théorique normale exprimée en Zscore (un Zscore étant une valeur représentant une différence par rapport à une population donnée). Les résultats de cette étude montrent qu'une différence de 1,96 entre la valeur théorique et la valeur réellement mesurée au niveau des sinus de valsava, peut refléter une dilatation aortique. Le simulateur de Zscore est accessible sur le site de la fondation Marfan http://www.marfan.org/dx/Zscore_
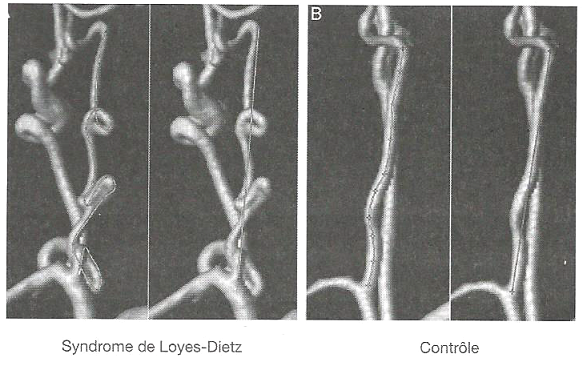
La tortuosité de l'artère vertébrale comme nouveau critère prédictif d'accidents cardiovasculaires
L'index de tortuosité de l'artère vertébrale calculé par imagerie est proposé comme un critère permettant d'évaluer l'apparition d'accidents cardiovasculaires.
Cette étude, réalisée sur une population âgée de moins de 50 ans, démontre qu'une augmentation de la tortuosité de l'artère vertébrale est associée à une dissection précoce de l'aorte thoracique.
L'augmentation de la tortuosité est retrouvée principalement chez les patients atteints des syndromes de Marfan et de Loeys-Dietz, et représente un critère permettant de prédire les accidents cardiovasculaires.
Caractérisation de la survenue de problèmes cardiaques chez des patients atteints du syndrome de Marfan et diagnostiqués durant l'enfance
Cette étude a été réalisée à partir de 465 patients atteints du syndrome de Marfan et diagnostiqués avant l'âge de 18 ans. Elle avait pour objectif de mieux comprendre l'occurrence d'apparition des problèmes cardiaques dans cette population. Un évènement cardiovasculaire est survenu chez 25 patients (5,4%), et parmi ceux-ci : 4,3% ont reçu une chirurgie prophylactique de l'aorte, 0,6% ont eu une dissection aortique et 0,4% sont décédés. Une intervention chirurgicale a été réalisée sur 23 patients (4,9%) dont le remplacement de la racine aortique par la procédure de Bentall avec valve mécanique (43,5%) ou avec conservation de la valve aortique (56,5%), et greffe coronarienne (17,4%). La courbe de survie Kaplan-Meir indique que 95% des patients n'ont subi aucun évènement cardio-vasculaire jusqu'à l'âge de 18 ans, et 78% jusqu'à l'âge de 30 ans. La chirurgie prophylactique réalisée sur aorte dilatée est la principale cause de survenue d'évènements cardiaques chez les patients atteints du syndrome de Marfan et diagnostiquée durant l'enfance.
Estimation du risque de complication cardiovasculaire dans le syndrome de Marfan
Cette étude a permis de démontrer que les patients atteints du syndrome de Marfan ont 156 fois plus de risque d'avoir une dissection aortique que la population générale (risque relatif = 156). Le risque pour les femmes atteintes du syndrome de Marfan est beaucoup plus élevé que pour les hommes (risque relatif pour les femmes = 264, contre 151 chez les hommes). Le risque d'anévrisme de l'aorte abdominale est également significativement augmenté (risque relatif = 10,51), tout comme le risque d'attaque cérébrale (risque relatif = 3,73).
Session 6 : Problèmes non cardiovasculaires et management dans le syndrome de Marfan et Apparentés
Management chirurgical et médical durant la grossesse et l’accouchement dans le syndrome de Marfan (Barbara Mulder)
Durant la grossesse des patientes atteintes du syndrome de Marfan des changements au niveau cardio-vasculaire peuvent avoir lieu et induire des tensions au niveau de la paroi aortique. Chez ces femmes, la grossesse entraîne une augmentation du risque de dissection aortique de l'ordre de 1%, associée à une forte mortalité fœtale et maternelle. La grossesse semble bien tolérée lorsque le diamètre de l'aorte ne dépasse pas 45 mm, cependant le risque de dissection aortique augmente avec le diamètre de l'aorte. Il est souvent recommandé de remplacer chirurgicalement la racine aortique en prévention lorsqu'une grossesse est désirée, puis de suivre le diamètre de l'aorte durant la grossesse et 6 mois après l’accouchement. En général, l’accouchement par voie naturelle est relativement sans danger sauf pour les femmes à haut risque cardiovasculaire pour lesquelles la césarienne est recommandée.
Rôle de l'oxytocine dans l’augmentation du risque de dissection aortique lors de la grossesse dans le syndrome de Marfan (Jennifer Habashi)
Cette étude s’intéresse au rôle de l'oxytocine dont la synthèse est augmentée en fin de grossesse et durant la lactation. Cette étude a été réalisée à partir d'expérimentation animale du syndrome de Marfan où l'incidence de la dissection aortique 4 semaines après la mise bas est de 91%. Ils ont montré que la séparation des bébés et de la mère afin d’éviter la production d'oxytocine durant la lactation permet de diminuer la mortalité avec une incidence de 26%.
Le traitement de ces souris avec un inhibiteur de l'oxytocine réduit le nombre de dissection aortique avec une occurrence de 6,7%. Ces travaux ouvrent une perspective thérapeutique dans le but de diminuer le risque de dissection aortique lors de la grossesse.
© Copyright ASFMa
Avec l’accord de Patrice Touboulie
Président AFSMa
- 503 views